Die starke gesetzliche Regulierung bei Medizinprodukten und der Healthcare-Branche, sowie ihre kontinuierliche Veränderung können durchaus Herausforderungen darstellen. Um in diesem kompetitiven Markt wettbewerbsfähig zu bleiben und zukunftsfähige, normenkonforme, sowie profitable Produkte herauszubringen, sind fundierte Kenntnisse der gesetzlichen Regelungen und gängigen Normen von großer Wichtigkeit. Durch unsere Unterstützung im Bereich Regulatory Affairs bieten wir Ihnen aktuellstes Wissen zu Gesetzen, Standards und Normen. Auch haben Sie die Möglichkeit, sich auf Ihre Kernkompetenz zu fokussieren.
Wir von senetics helfen Ihnen dabei, die Normen und Regularien in Ihrem Unternehmen einzuführen und auf dem neuesten Stand zu bleiben.
Bitte beachten Sie:
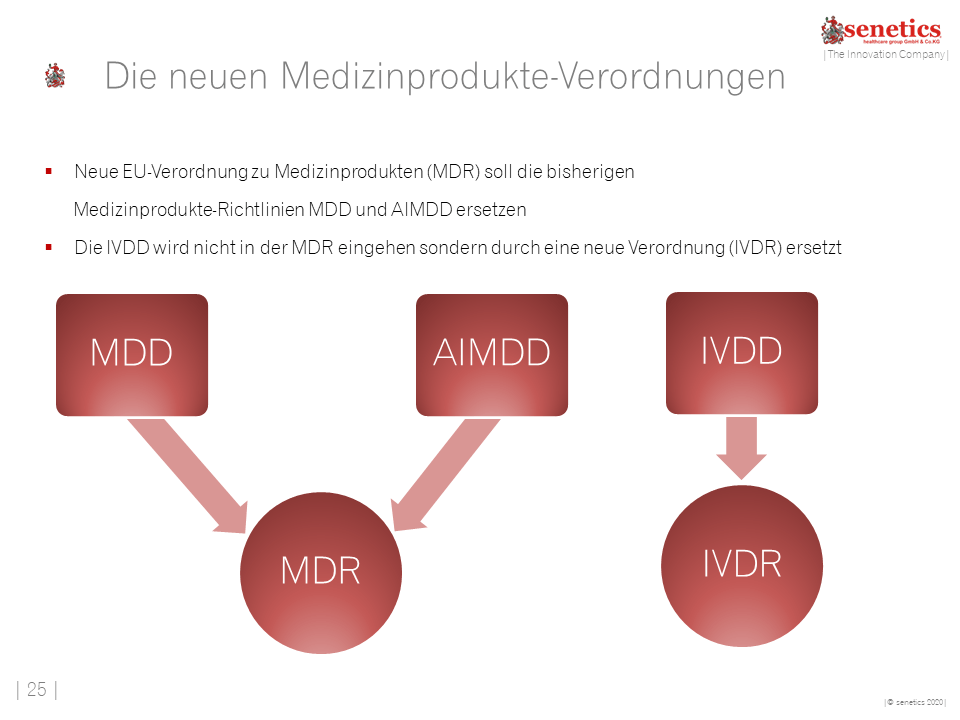
- Für In-vitro Diagnostik wurden die Übergangsfristen verlängert. Die IVDR gilt ab 26.05.2022
Bereiten Sie Ihr Unternehmen rechtzeitig auf diese Umstellung vor!
Was ist ein Medizinprodukt, was muss man beachten?
Was unter dem Begriff Medizinprodukt zu verstehen ist, steht im § 3 des Medizinproduktegesetzes (MPG).
1) Als Medizinprodukt werden alle „(…) Instrumente, Apparate, Vorrichtungen, Software, Stoffe oder anderen Gegenstände (…)“ bezeichnet, die vom Hersteller zur Anwendung am Menschen für folgende Zwecke bestimmt sind:
-
Erkennung, Verhütung, Überwachung, Behandlung oder Linderung von Krankheiten
-
Erkennung, Überwachung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen
-
Untersuchung, Ersatz oder Veränderung des anatomischen Aufbaus oder eines physiologischen Vorgangs
-
Empfängnisregelung, mit „(…) Hauptwirkung im oder am menschlichen Körper weder durch pharmakologisch oder immunologisch wirkende Mittel noch durch Metabolismus erreicht wird, deren Wirkungsweise aber durch solche Mittel unterstützt werden kann.“
Sie fragen sich ob Ihr Produkt unter den Geltungsbereich des Medizinproduktegesetzes fällt? Dies legt der Hersteller des Medizinproduktes in der Zweckbestimmung fest. Gerne unterstützen wir Sie bei dieser und allen anderen Fragestellungen.

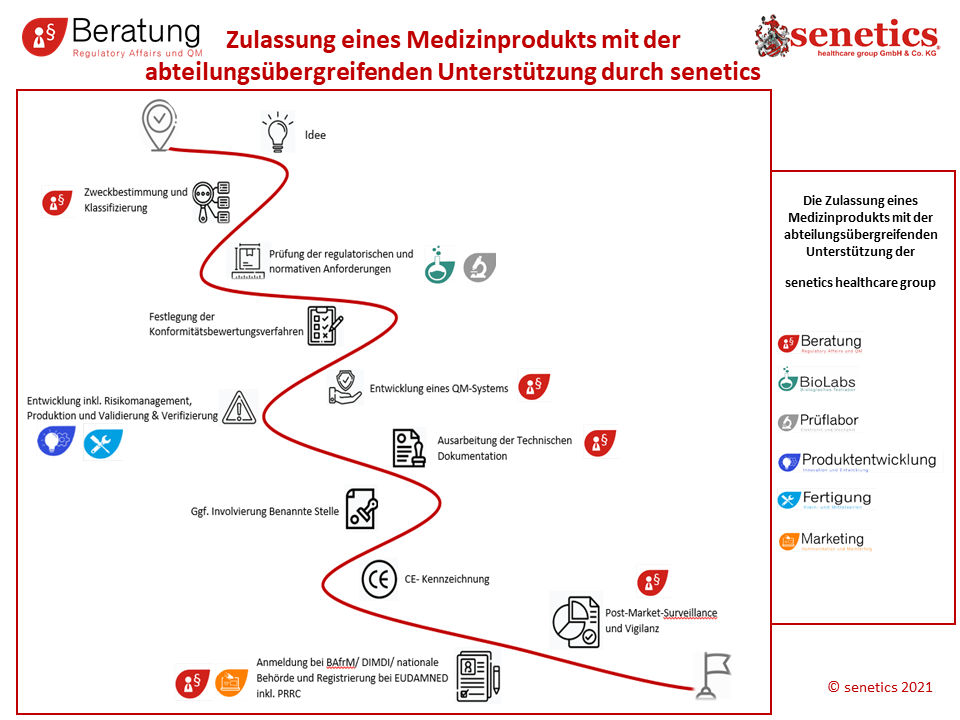
Wir unterstützen Sie gerne bei folgenden Herausforderungen:
- Zulassung EU nach den „neuen“ Verordnungen
- Umsetzung der neuen MDR und IVDR im laufenden Betrieb
- Implementierung eines Qualitätsmanagementsystems nach DIN EN ISO 13485
- Konformitätsbewertungsverfahren für Ihr Produkt
- Erstellen der Technischen Dokumentation nach aktuellen Anforderungen:
- Erstellung und Pflege der Technischen Dokumentation nach MDR, IVDR
- Überarbeitung Ihrer Technischen Dokumentation nach Auditabweichungen
- Klinische Bewertungen nach MEDDEV 2.7/1 Rev. 4 (MDD/MDR)
- Klinische Prüfungen nach DIN EN ISO 14155
- Stellung der „Verantwortlichen Person für regulatorische Anforderungen“ nach EU-Verordnung 2017/745 (MDR) (Art. 15)
- Stellung des „Sicherheitsbeauftragten“ nach §30 MPG (nur für IVD)
- Zulassung Ihres Produktes in den USA nach
- FDA 510(k) Premarket Notification
- FDA Premarket Approval (PMA)
Was ist eine Zweckbestimmung für Medizinprodukte?
Die Zweckbestimmung ist Teil der gesetzlichen Auflagen (Regulatory Affairs) und zwingend notwendig für die erfolgreiche Zulassung eines Medizinproduktes. Die Zweckbestimmung ist der 1. Schritt und die Grundlage für die Zuordnung zur Kategorie Medizinprodukte, ebenso wie zur Klassifizierung des Medizinproduktes. Diese basiert im Wesentlichen auf den Fragen:
- Was ist der medizinische Zweck des Produkts?
- Wer sind die vorgesehenen Nutzer- und Patientengruppen?
- Für welche Nutzungsbereiche, medizinische Indikationen und Körperbereiche ist das Produkt vorgesehen?
- Welchen Funktionsprinzipien und Wirkungsmechanismen besitzt das Produkt?
Auf Basis der Zweckbestimmung erfolgt die Risikoklassifizierung. Im Anschluss kann das Konformitätsbewertungsverfahren mit anschließender Zulassung abgeleitet werden.
Mit senetics sind Sie auf der sicheren Seite.
Kommen Sie gerne auf uns zu, wir unterstützen Sie bei Ihrem Anliegen!