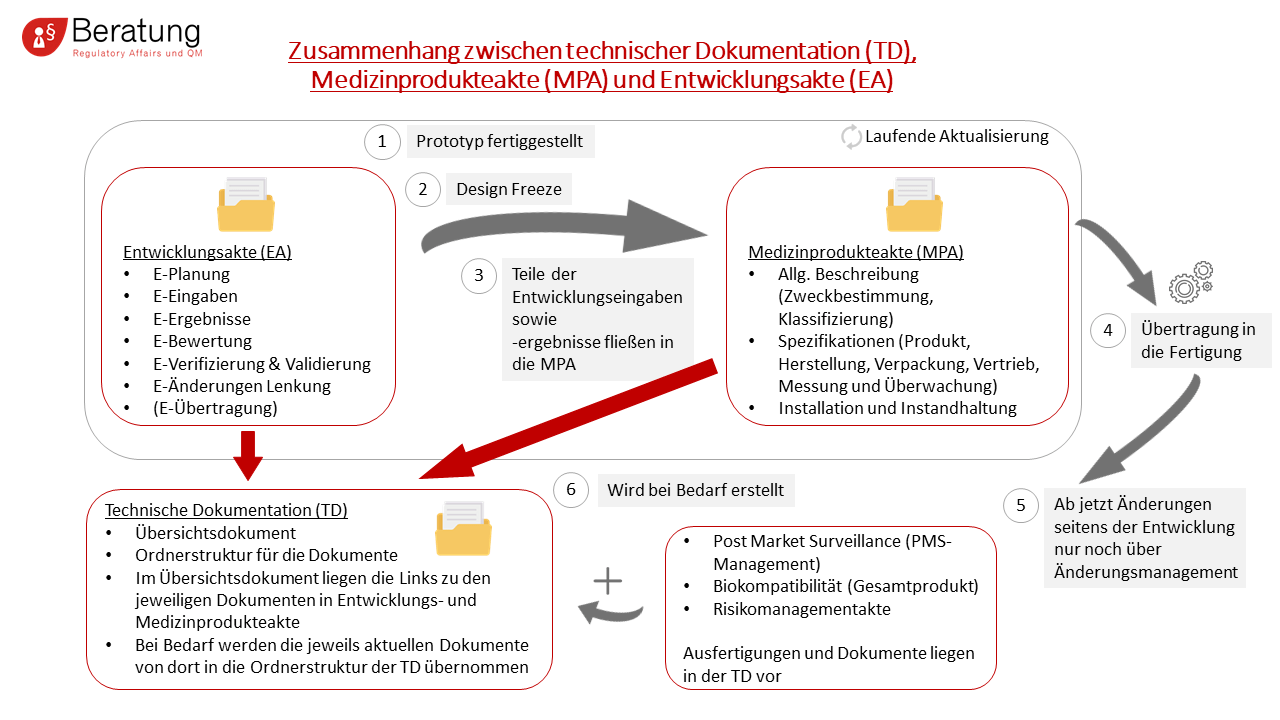
Was ist die technische Dokumentation eines Medizinproduktes?
Eine saubere technische Dokumentation gemäß Medical Device Regulation MDR bzw. In-vitro-Diagnostic Device Regulation IVDR ist die Grundvoraussetzung für die Erteilung einer Zulassung. Diese umfasst alle Dokumente und Aufzeichnungen zu Entwicklung, Produktion, Marketing und Instandhaltung Ihres technischen Erzeugnisses. Ihr Aufbau und ihre Struktur müssen so gestaltet sein, dass der jeweilige Zweck gemäß MDR bzw. IVDR vollständig erfüllt wird. Die technische Dokumentation dient Ihrem Unternehmen als Nachweis, dass Sie die Grundlegenden Anforderungen aus Anhang I der MDD (93/42/EWG) oder Anhang II der MDR und IVDR erfüllen. Der Zweck der technischen Dokumentation ist die Information und Instruktion der Nutzer, die haftungsrechtliche Absicherung des Herstellers, die Produktbeobachtung, die Rückverfolgbarkeit und Reproduzierbarkeit, sowie die Archivierung der Informationen.
Technische Dokumentation – Regulatorische Vorgaben
- Zulassung EU nach den „neuen“ Verordnungen (bitte beachten Sie die Übergangsfristen!)
- Zulassung EU nach den „alter“ Richtlinie (Diese gilt bis 26.05.2022)
- IVDD (98/79/EG)
- Zulassung Ihres Produktes in den USA nach FDA 510(k) Premarket Notification
- FDA Premarket Approval (PMA)
Technische Dokumentation – Normative Vorgaben
Die normativen Anforderungen für die technische Dokumentation bei Medizinprodukten und in der Medizintechnik sind vielfältig. Zudem variieren die Vorgaben je nach dem Anwendungsbereich und den Leistungsmerkmalen des Medizinproduktes. Die Anwendung von harmonisierten Normen für die Dokumentation von Medizinprodukten, ist ein wesentlicher Teil, um Konformität nachzuweisen.
- EN ISO 13485 (Qualitätsmanagementsysteme für Medizinprodukte)
- ISO 14971 (Risikomanagement für Medizinprodukte)
- EN 60601 (Medizinisch elektrische Geräte)
- IEC 62366–1 (Anwendung der Gebrauchstauglichkeit auf Medizinprodukte)
- IEC 62304 (Software für medizinische Geräte)IEC/IEEE 82079–1 (Erstellen von Nutzerinformationen)
- ISO 17664 (Herstellerangaben für Medizinprodukte)
- EN ISO 8536–4 (Infusionsgeräte zur medizinischen Verwendung)
- ISO 14937 (Sterilisation von Medizinprodukten)
- ISO 20417 (Medizinprodukte — Allgemeine Informationen des Herstellers)
- EN 1041 (Bereitstellung von Informationen durch den Hersteller von Medizinprodukten)
- ISO 15223–1 (Bei Aufschriften von Medizinprodukten zu verwendende Symbole)
- IEC 62366–2 (Leitfaden zur Anwendung des Usability Engineering auf Medizinprodukte)
- u.s.w.
International Medical Device Regulators Forum (IMDRF) und “Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical Devices (STED)“.
Das International Medical Device Regulators Forum (IMDRF – ehemals Global Harmonization Task Force – GHTF) hat final im März 2019 einen Leitfaden veröffentlicht: „Non-In Vitro Diagnostic Device Market Authorization Table of Contents“ (nIVD MA ToC). Ergebnis war die „Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical Devices (STED)“. Die Medizinprodukte-Verordnung (MDR) verweist explizit auf die Arbeit dieses Ausschusses als Grundlage für das Erstellen der Technischen Dokumentation von Medizinprodukten. Für In-vitro Diagnostika gibt es eine entsprechende Vorlage.
STED (Summary Technical Documentation)
a) Übersicht
STED strukturiert die technische Dokumentation in über 200 Kapiteln und Unterkapiteln:
- Marktspezifische und regulatorische Dokumente
- Beschreibung des Produkts
- Nicht klinische Nachweise
- Klinische Nachweise
- Labeling
- QM-System: Verfahren und Methoden
- QM-System: Produktspezifische Vorgaben
STED in der Praxis
Die Struktur der STED (Summary Technical Documentation) für Medizinprodukte ist mit über 200 Kapiteln und Unterkapiteln sehr umfangreich. Allerdings ist dies für Hersteller eine wichtige Strukturierung. Die Summary Technical Documentation ist ein wertvolles Hilfsmittel, um die technische Dokumentation zu strukturieren. Es gibt auch mögliche alternative Strukturen, die sich ebenso eignen.
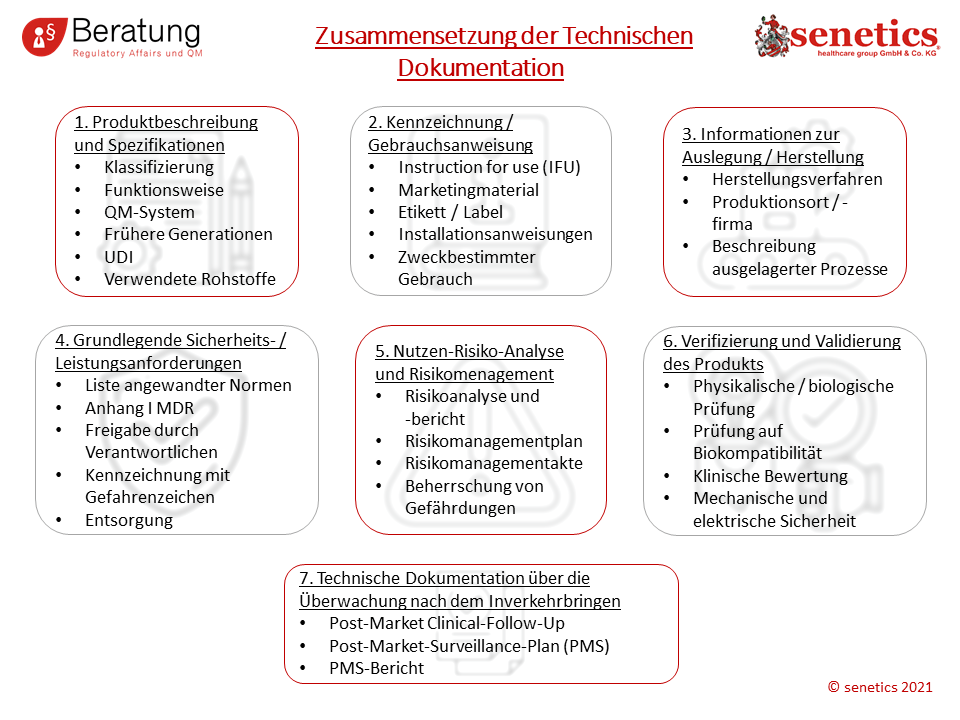
Nach Anhang II MDR ist (in Kurzform) diese Struktur gefordert:
- Eine Produktbeschreibung und Spezifikation mit allen Varianten und Zubehörteilen (Beschreibung des Produktes, Spezifikation und Angaben zum Produkt wie dessen Name, Unique Device Identification (UDI) , Klassifizierung, Hinweis auf frühere Produkte)
- Auflistung aller Teile, die vom Hersteller mitgeliefert werden müssen
- Herstellungsinformationen des Produktes
- Die Grundlegende Sicherheits- und Leistungsanforderungen (Essential Principles)
- Risikomanagement mit entsprechenden Analysen
- Verifizierung und Validierung des Produktes (mit Klinischen Daten und Vorklinischen Daten (entsprechend Anhang XIV MDR, Informationen zu z.B. Stoffen, Gewebe und Zellen menschlichen oder tierischen Ursprungs, und Produktionsangaben bei sterilen Produkten etc.)
Klinische Bewertung von Medizinprodukten als Teil der Technischen Dokumentation
Die Klinische Bewertung ist die Analyse und Bewertung von klinischen Daten zu einem spezifischen Medizinprodukt mit dem Ziel, dessen Sicherheit, Eignung und Leistung in der klinischen Anwendung nachzuweisen. Sie erfolgt anhand von klinischen Daten von z.B. klinischen Prüfungen und/oder auf Basis wissenschaftlicher Literatur.
Die klinische Bewertung ist in Europa Teil des Konformitätsbewertungsverfahrens für Medizinprodukte (CE-Kennzeichnung) und bei allen Medizinprodukten notwendig. Sie ist abzugrenzen von einer Klinischen Studie/Klinischen Prüfung.
Unsere Dienste
Wir unterstützen Sie bei der Erstellung der Technischen Dokumentation unter Berücksichtigung der MDR. Dabei können wir Sie bei der Erstellung der Technischen Dokumentation beraten, als auch die Erstellung einzelner Dokumente, wie der Risikomanagementakte oder Usability-Engineering-Akte, für Sie übernehmen. Somit erhalten Sie perfekte Dokumente, welche Sie in den folgenden Zulassungen als Vorlage verwenden können, sodass Sie sich ganz auf Ihre Kern-Kompetenzen konzentrieren können.
– komplette Erstellung der Technischen Dokumentation
– Hilfestellung/ Beratung bei Ihrer Technischen Dokumentation
– Überprüfung Ihrer Technischen Dokumentation hinsichtlich der notwendigen Anforderungen für eine Zulassung
– Überarbeitung Ihrer Technischen Dokumentation nach Auditabweichungen
Prüfung ihrer vorhandenen technischen Dokumentation
Eine Technische Dokumentation muss alle erforderlichen Anforderungen beinhalten, damit die Zulassung für das Produkt erteilt wird. Ist diese unvollständig, oder nicht normenkonform, hat dies einen unnötigen Verwaltungsaufwand zur Folge. Daher bieten wir Ihnen eine Überprüfung Ihrer Technischen Dokumentation anhand aller wichtigen und notwendigen Gesichtspunkte an.
Wir prüfen für Sie:
– Software-Lebenszyklus gemäß IEC 62304
– Einhaltung der grundlegenden Anforderungen nach MPG und MDR
– Risikomanagementakte
– Usability-Management-Akte
Mit senetics sind Sie auf der sicheren Seite.
Kommen Sie gerne auf uns zu, wir unterstützen Sie bei Ihrem Anliegen!