EU-Zulassung
Medizinprodukte, die im Europäischen Wirtschaftsraum (EWR) erstmalig in Verkehr gebracht werden sollen, benötigen, mit Ausnahme von Sonderanfertigungen, eine CE-Kennzeichnung. Dies betrifft sowohl Medizinprodukte sowie In-Vitro Diagnostika (IVD). Das Konformitätsbewertungsverfahren mit dem Ziel, das CE-Zeichen zu erhalten, müssen die Hersteller selbst durchlaufen. Durch das CE-Zeichen versichert der Hersteller, dass sein Produkt die grundlegenden Anforderungen erfüllt.
Den rechtlichen Rahmen bilden in Europa die Verordnungen (EU) 2017/745 und 2017/746 (MDR & IVDR).
Die senetics healthcare group unterstützt Sie bei der Zulassung Ihres Produkts umfangreich in folgenden Bereichen:
- Bestimmung der Risikoklasse
- Ausarbeitung einer geeigneten Zulassungsstrategie
- Feststellung anzuwendender (harmonisierter) Normen und Standards
- Vorbereitung der Technischen Dokumentation
- Umsetzung eines ISO 13485 Qualitätssicherungssystems und Auditierung
- Beratung zur Durchführung des Risikomanagements nach ISO 14971
- Unterstützung bei der Kennzeichnung von Medizinprodukten
- Durchführung der klinischen Bewertung
- Beratung zur Durchführung einer klinischen Prüfung, wenn diese notwendig ist
- Auswahl und Kommunikation mit der benannten Stelle
- Unterstützung in der Marktbeobachtung und beim Aufbau eines Vigilanzsystems / Stellung der verantwortlichen Person (PRRC)
Wie kann man die Risikoklasse eines Medizinproduktes bestimmen?
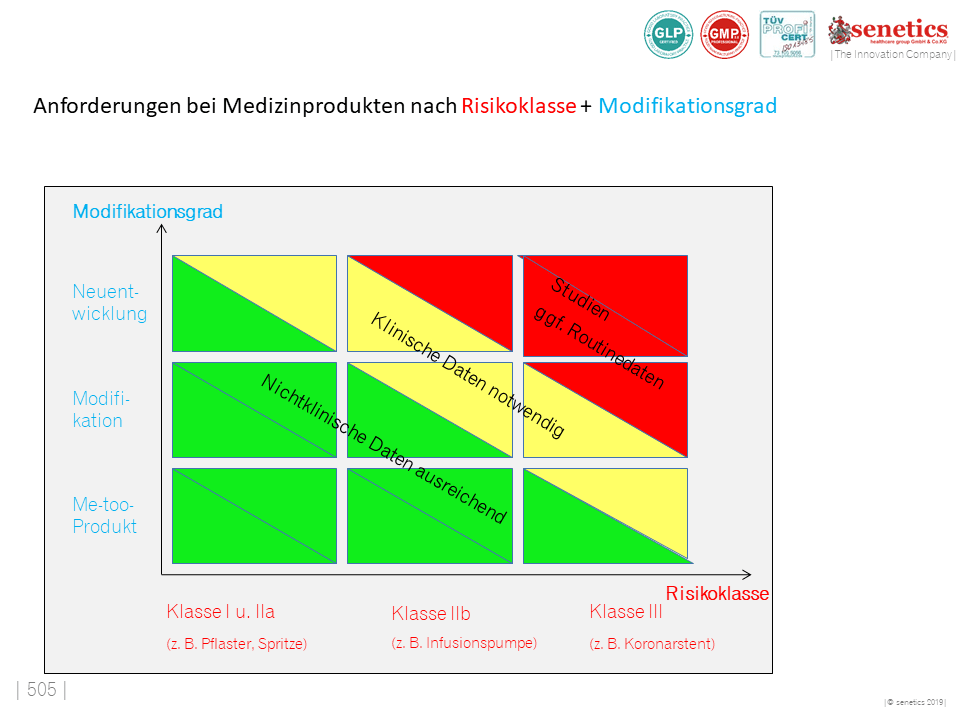
Medizinprodukte werden Risikoklassen zugeordnet. Der Hersteller ist dafür verantwortlich, sein Medizinprodukt anhand vordefinierter Kriterien einer der nachfolgenden Klassen zuzuordnen. Die Klassifizierung des Medizinprodukts steht dabei in direktem Zusammenhang mit dem Risiko in Bezug zum Patienten, Anwender und Dritten.
Die Klassifizierung erfolgt nach den Klassifizierungsregeln des Anhangs VIII der Verordnung 2017/745 (MDR). Die Produkte werden in die vier Klassen I, IIa, IIb und III unterteilt.
Die regulatorischen Vorgaben formulieren definierte Bewertungsmaßstäbe, anhand derer die Hersteller von Medizinprodukten eine zuverlässige Klassifizierung vornehmen können. Eine inkorrekte oder falsche Zuordnung kann nicht nur erhebliche finanzielle Risiken bergen, sondern auch dazu führen, dass es Schwierigkeiten mit dem Gesetzgeber und Behörden gibt. Gerne unterstützen wir Sie bei der korrekten Risikoklassifizierung.
Unsere Experten unterstützen Sie weiterhin von der Idee bis zur fertigen Konformitätserklärung in allen Bereichen.
Mit senetics sind Sie auf der sicheren Seite.
Kommen Sie gerne auf uns zu, wir unterstützen Sie bei Ihrem Anliegen!