Seit dem 21.05.2021 gilt offiziell die MDR als Nachfolger der MDD. Die Verordnung enthält die rechtlichen Anforderungen an das erstmalige Inverkehrbringen von Medizinprodukten im Europäischen Wirtschaftsraum.
Die Medical Device Regulation bringt einige umfangreiche Änderungen mit sich, u. a.:
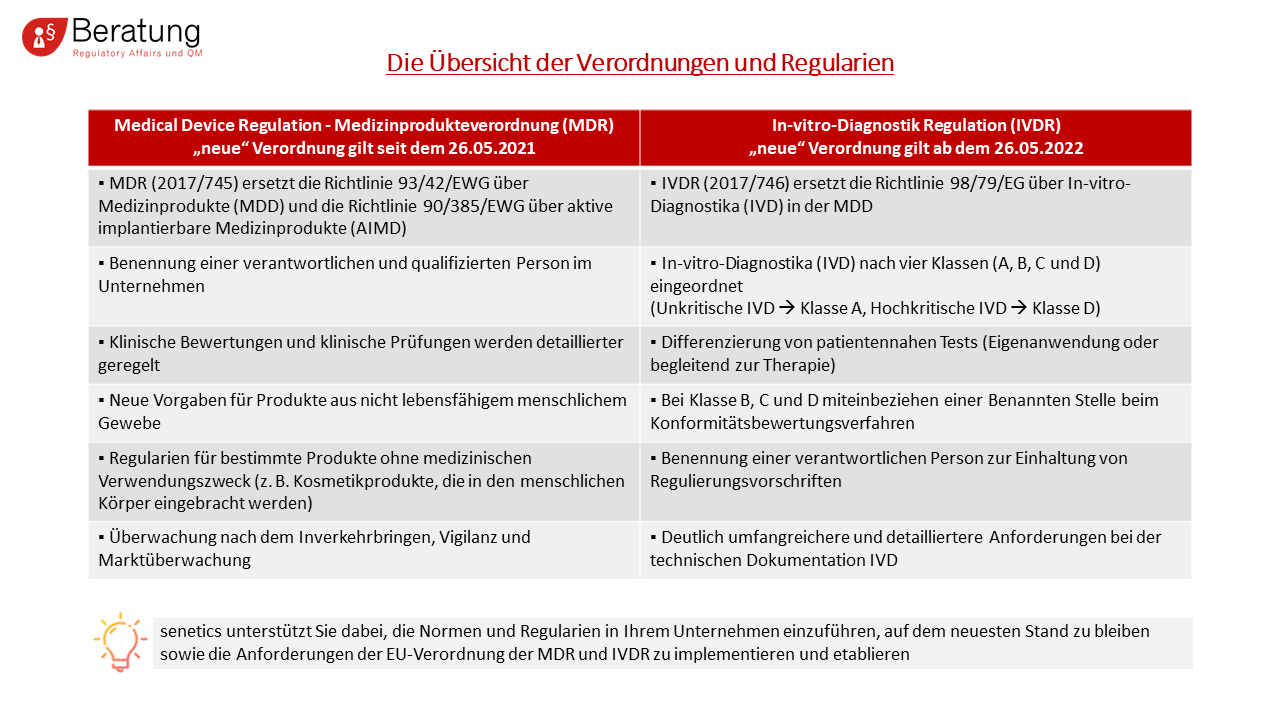
- Neues „scrutiny-Verfahren“ für Produkte mit hohem Risiko
- Inhalt und Umfang der technische Dokumentation werden detaillierter geregelt
- Alle Produkte müssen eine eindeutige Identifikationsnummer, die UDI, tragen
- Hersteller müssen eine verantwortliche/qualifizierte Person im Unternehmen benennen, die sich mit regulatorischen Fragestellungen auseinandersetzt
- Die europäische Datenbank EUDAMED wird ausgeweitet und soll zum zentralen Hub für Medizinprodukte werden
- Klinische Bewertungen und klinische Prüfungen werden detaillierter geregelt
- Unangekündigte Audits durch die benannten Stellen werden zunehmen
- Die Anforderungen an die Post-Market Surveillance steigen und sind detaillierter geregelt
- Umfangreiche Änderungen für OEM-Hersteller

Anforderungen der Medical Device Regulation
Das Inverkehrbringen eines Medizinproduktes ist nur dann zulässig, wenn es den nötigen Anforderungen der MDR, wie z.B. der Zweckbestimmung oder der Instandhaltung entspricht.
Zudem arbeiten die Händler und Importeure mit den Herstellern oder ihren Bevollmächtigten zur Erreichung eines angemessenem Niveaus der Rückverfolgbarkeit von Produkten zusammen.
Bewertungsverfahren
Jedes Medizinprodukt, welches in der EU auf den Markt kommt, muss gekennzeichnet sein. Dies stellt sicher, dass die gestellten Anforderungen an das Produkt erfüllt sind. Um solch ein CE-Kennzeichen zu erhalten, muss der Hersteller ein Konformitätsbewertungsverfahren durchlaufen. Auch die Risikoklasse des Produktes – mit Ausnahme von in-vitro-Diagnostika und aktiven implantierbaren Medizinprodukten – muss bestimmt sein. Die Konformitätsbewertung erfolgt über die sogenannte Benannte Stelle. Diese muss bestimmte Anforderungen erfüllen.
Zusätzlich ist eine Klinische Bewertung und gegebenenfalls eine Klinische Prüfung bindend.
Überwachung nach Inverkehrbringen, Vigilanz und Martküberwachung
Nach der Veröffentlichung des Medizinprodukts werden aktiv und systematisch Informationen zur Produktverwendung im Markt gesammelt. Auf diese Weise werden Vorkommnisse und Trends ermittelt. Falls Korrektur- und/oder Präventivmaßnahmen erforderlich sind, unterrichtet der Hersteller die Anwender, zuständigen Behörden oder die Benannte Stelle.
Die Vigilanz ist ein gesondert geregeltes Verfahren. Hier stellt der Hersteller ein schwerwiegendes Vorkommnis fest und leitet daraufhin eine Sicherheitskorrekturmaßnahme ein, welche unverzüglich zu melden ist.
Kooperation zwischen den Mitgliedstaaten
Die Mitgliedstaaten benennen die für die Durchführung dieser Verordnung zuständigen Behörden und statten diese mit den notwendigen Befugnissen, Ressourcen, Ausrüstungen und Kenntnissen aus. Durch die Kooperation zwischen den zuständigen Behörden der Mitgliedstaaten und der Kommission ist ein organisierter Informationsaustausch möglich.
Register und Datenbanken sind für die unabhängige Bewertung der langfristigen Sicherheit und Leistung der Produkte oder der Rückverfolgbarkeit implantierbarer Produkte essenziell.
Vertraulichkeit, Datenschutz, Finanzierung und Sanktionen
Die im Rahmen der Durchführung erlangten Informationen und Daten sind vertraulich zu wahren. Dies gilt für alle an der Anwendung beteiligten Personen, sofern es in der Verordnung nicht anders vorgesehen ist. Hierbei ist die nationale Gesetzgebung zu beachten.
Mit senetics sind Sie auf der sicheren Seite.
Kommen Sie gerne auf uns zu, wir unterstützen Sie bei Ihrem Anliegen!